Why the interest in CLL and the ATM gene?
When doing research on Chronic Lymphocytic Leukemia, I focused mainly on those patients that had an 11q- (see homepage). These patients were especially intriguing due to the fact that they tend to experience more aggressive disease and, therefore, a poorer prognosis when compared to most other CLL patients [1]. One of the possible contributors to this more aggressive disease is the occurrence of chemoresistance (reduced sensitivity to chemotherapy) in leukemic cells lacking both copies of a crucial gene deleted in an 11q-, ATM [2]. If the chemotherapy is not effective in reduction in the leukemic cell line, it allows for the cancer to continue to grow and develop which will then lead to a reduction in the overall survival of the patient. During my research, I focused extensively on trying to find an explanation as to why some of these patients experience this chemoresistance.
Ataxia Telangiectasia Mutated (ATM), a gene deleted in this genetic abnormality, has been the focus of multiple studies and was also the gene of interest for my project. Given its role in DNA repair, it is no surprise that this gene is important in the development and progression of cancer. The CLL patients with the 11q- genetic abnormality still have one good copy of the gene, which has been shown to be sufficient in maintaining normal cellular function [3]. The problem is that these patients are also at higher risk of developing a mutation at that one good allele, which is when the problems arise. When the leukemic cells become ATM-/-, they become less sensitive to the chemotherapy drugs. [1]. With that said, I purpose future experiments that can be done to further look into why leukemic cells lacking a good copy of ATM experience this resistance to death, which can help explain why CLL patients with an 11q- experience a poorer clinical outcome.
Ataxia Telangiectasia Mutated (ATM), a gene deleted in this genetic abnormality, has been the focus of multiple studies and was also the gene of interest for my project. Given its role in DNA repair, it is no surprise that this gene is important in the development and progression of cancer. The CLL patients with the 11q- genetic abnormality still have one good copy of the gene, which has been shown to be sufficient in maintaining normal cellular function [3]. The problem is that these patients are also at higher risk of developing a mutation at that one good allele, which is when the problems arise. When the leukemic cells become ATM-/-, they become less sensitive to the chemotherapy drugs. [1]. With that said, I purpose future experiments that can be done to further look into why leukemic cells lacking a good copy of ATM experience this resistance to death, which can help explain why CLL patients with an 11q- experience a poorer clinical outcome.
proposed Experiments:
When studying ATM’s interaction network, there was one protein that looked to be of interest as it might potentially help in explaining the poorer prognosis seen in CLL patients that harbor an 11q-. This protein, named Sirtuin 1 (SIRT1), is known to be a deacetylase which means it regulates its substrates by removing an acetyl group from them, rendering them inactive. One important substrate of SIRT1 is p53, which, like ATM, is important in maintaining the health of the cell by playing a role in cell cycle arrest as well as apoptosis (cell death) upon the occurrence of DNA damage. This interaction was one of the reasons for my interest in SIRT1 as p53 is associated with a variety of cancers. Sirtuin 1 has been shown to regulated p53 by inactivating it when it is not needed within the cell [4].
Another important role of SIRT1 is its role in regulating autophagy, which is a process the cell uses to recycle its damaged/unwanted components. It has been shown that elevated levels of SIRT1 lead to an increase in the amount of autophagy, allowing the cell the live longer [5]. Consequently, it has been shown that increased levels of autophagy can also lead to survival of cancerous cells in the face of genotoxic stress induced by chemotherapy [6].
Given SIRT1’s role in regulating p53 and inducing autophagy, it can be speculated that increased levels of SIRT1 leads to an increase in cellular survival. Knowing this, it is important to understand how ATM regulates SIRT1, which can then lead to possible answers as to why CLL patients who are deficient in ATM experience a more aggressive cancer. Research has shown that ATM regulates SIRT1, indirectly, through the phosphorylation of another protein, DBC1. When DBC1 becomes phosphorylated, this leads to the inactivation of SIRT1. Therefore, a higher level of active ATM leads to lower levels of active SIRT1 and vice versa (figure 2) [7]. With this in mind, I wanted to study cells that were ATM deficient and examine the levels of active SIRT1 to see how it differed from cells that have at least one copy of ATM (ATM +/+, ATM+/-).
Another important role of SIRT1 is its role in regulating autophagy, which is a process the cell uses to recycle its damaged/unwanted components. It has been shown that elevated levels of SIRT1 lead to an increase in the amount of autophagy, allowing the cell the live longer [5]. Consequently, it has been shown that increased levels of autophagy can also lead to survival of cancerous cells in the face of genotoxic stress induced by chemotherapy [6].
Given SIRT1’s role in regulating p53 and inducing autophagy, it can be speculated that increased levels of SIRT1 leads to an increase in cellular survival. Knowing this, it is important to understand how ATM regulates SIRT1, which can then lead to possible answers as to why CLL patients who are deficient in ATM experience a more aggressive cancer. Research has shown that ATM regulates SIRT1, indirectly, through the phosphorylation of another protein, DBC1. When DBC1 becomes phosphorylated, this leads to the inactivation of SIRT1. Therefore, a higher level of active ATM leads to lower levels of active SIRT1 and vice versa (figure 2) [7]. With this in mind, I wanted to study cells that were ATM deficient and examine the levels of active SIRT1 to see how it differed from cells that have at least one copy of ATM (ATM +/+, ATM+/-).
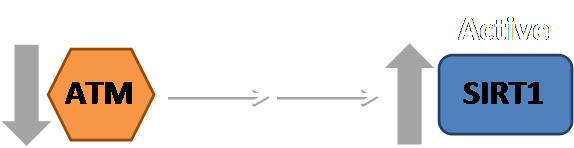
Figure 1: Lower levels of active ATM presumably leads to higher levels of active Sirtuin 1 (SIRT1). The double white arrows signifies that ATM does not directly interact with SIRT1. Instead, ATM regulates it through the phosphorylation of another protein, DBC1. This phosphorylation leads to the inactivation of SIRT1.
Experiment #1:
Question: How are levels of active SIRT1 affected in ATM +/- and ATM -/- cells?
Hypothesis: There will be higher levels of active SIRT1 in ATM -/- but the levels of active SIRT1 in ATM +/- cells will be comparable to wild type cells (ATM +/+).
Model Organism: Mice/Humans with CLL and an 11q-.
Method: Do a 2-D gel on leukemic cells that are ATM+/- and ATM -/- while using a healthy B-cell (ATM+/+) as the control. (Figure 2)
Hypothesis: There will be higher levels of active SIRT1 in ATM -/- but the levels of active SIRT1 in ATM +/- cells will be comparable to wild type cells (ATM +/+).
Model Organism: Mice/Humans with CLL and an 11q-.
Method: Do a 2-D gel on leukemic cells that are ATM+/- and ATM -/- while using a healthy B-cell (ATM+/+) as the control. (Figure 2)
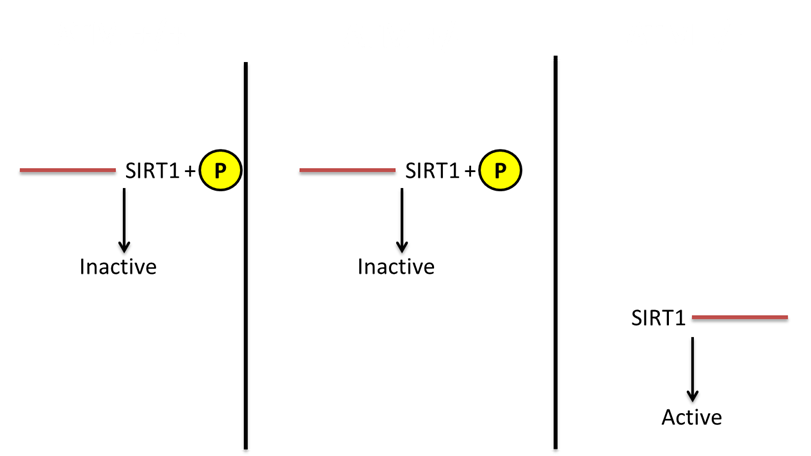
Figure 2: The expected 2-D gel from the proposed experiment. As described, these proteins would be isolated from B-cells with the respective genotype and assayed using standard 2-D gel protocols. This is a simplified version of what the expected gel in order to effectively show the expect results. The yellow P signifies that the presence of phosphorylation. It should be noted that this phosphorylation is present on the DBC1 protein, which SIRT1 binds to and becomes inactivated [7]. This phosphorylation adds more weight to the protein which is why it is higher up on the gel.
From this experiment, I would expect to find that in those cells that are ATM deficient (ATM-/-) higher levels of SIRT1 would be present. For those cells that still have one good copy of ATM, ATM +/-, SIRT1 levels would not be different than that of wild type cells because it has been shown that the levels of active ATM does not differ between ATM +/- cells and ATM +/+ cells [3]. Knowing this, it can be speculated that SIRT1 would be regulated appropriately in those cells possessing only one viable copy of ATM.
So why is this important? Well, as stated earlier and in the popular press article, those CLL patients with an 11q- are at higher risk for developing a subsequent ATM mutation at the other allele which would then render their cells ATM-/-. These cells will then not regulated SIRT1 correctly, which will allow these cancerous cells to survive longer due to the disruption of p53 regulation and the up-regulation in autophagy. With p53 and autophagy not being regulated properly in the leukemic cells, the patient could become desensitized to normal chemotherapeutics used for CLL patients due to the cancerous cells ability to avoid apoptosis. This is why it would beneficial to find a way to inhibit SIRT1 within the blood, so that these patients might become more sensitive to chemotherapeutics and, therefore, have a better outcome. This is the idea that led to the second proposed experiment.
So why is this important? Well, as stated earlier and in the popular press article, those CLL patients with an 11q- are at higher risk for developing a subsequent ATM mutation at the other allele which would then render their cells ATM-/-. These cells will then not regulated SIRT1 correctly, which will allow these cancerous cells to survive longer due to the disruption of p53 regulation and the up-regulation in autophagy. With p53 and autophagy not being regulated properly in the leukemic cells, the patient could become desensitized to normal chemotherapeutics used for CLL patients due to the cancerous cells ability to avoid apoptosis. This is why it would beneficial to find a way to inhibit SIRT1 within the blood, so that these patients might become more sensitive to chemotherapeutics and, therefore, have a better outcome. This is the idea that led to the second proposed experiment.
Experiment #2:
Question: Are there specific phosphorylation sites on SIRT1 that lead to its activation in the blood?
Hypothesis: Sirtuin 1 is regulated differently in the blood, possibly via phosphorylation.
Model Organism: Mice
Method: Conducted a bioinformatics search for phosphorylation sites on SIRT1 (figure 3) and aligned protein sequences of organisms with and without blood (figure 4).
Hypothesis: Sirtuin 1 is regulated differently in the blood, possibly via phosphorylation.
Model Organism: Mice
Method: Conducted a bioinformatics search for phosphorylation sites on SIRT1 (figure 3) and aligned protein sequences of organisms with and without blood (figure 4).
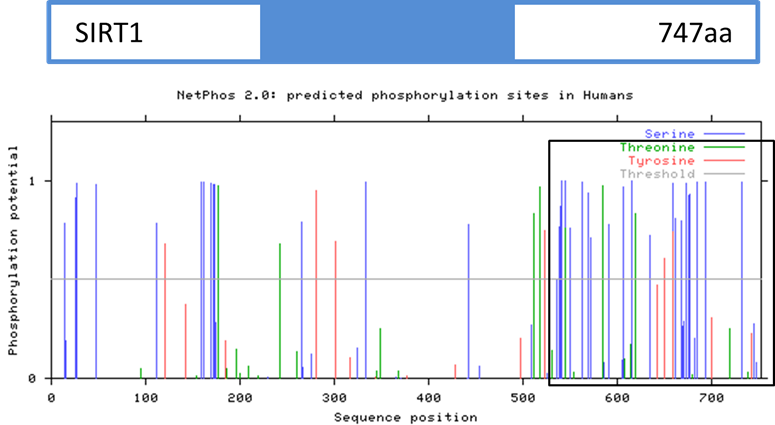
Figure 3: The protein sequence of SIRT1 was subject to NetPhos to determine possible phosphorylation sites. The black box indicates the C-terminus region of the SIRT1 where I further focused to find possible phosphorylation sites that could lead to SIRT1 activation in the blood. The grey line on the graph indicates the threshold which is used to determine the likelihood of a residue becoming phosphorylated. Those that are above the threshold have a greater chance at becoming phosphorylated. This is why I focused on the C-term region, as it has a plethora of residues that are likely to be phosphorylated.
In addition to the benefit that would come from finding a phosphorylation site that was specific to SIRT1 activity in the blood, another reason why I choose to ask this question is because SIRT1 is known to be a tumor suppressor as well as an oncogene (contributes to cancer if mutated) depending on the tissue it is found in [8]. For example, it has been shown in mice that the expression of SIRT1 in the intestine leads to reduced intestinal tumors, evidence for tumor suppressor function [9]. On the other hand, research has also found that the knocking out SIRT1 lead to impaired development of CML (Chronic Myelogenous Leukemia), which is evidence for SIRT1 function as an oncogene [10]. Furthermore, it was shown that mice that are deficient in SIRT1 show a lot of developmental defects and die during the postnatal period [4], alluding to the importance of SIRT1 in organism viability. Therefore, knocking out SIRT1 completely would result in more harm than good, which is why I wanted to focus on knocking it out solely in the blood.
In order to further look into this, the SIRT1 protein sequence was subjected to NetPhos, a site that predicts the residues (amino acids) that are most likely to become phosphorylated. Upon doing this, it was found that there were a multitude of residues that were likely to become phosphorylated at the C-terminus end of the protein, which is where I focused further to find conserved residues among different organisms with blood. Using ClustalW2, the SIRT1 protein sequences of various organisms, with and without blood, were aligned and it was found that there were three serine residues (denoted as S) that were conserved among organisms with blood, but were not found in organisms without (figure 4). Furthermore, when looking at the results from NetPhos, it was found that these three residues had a high likelihood of becoming phosphorylated.
In order to further look into this, the SIRT1 protein sequence was subjected to NetPhos, a site that predicts the residues (amino acids) that are most likely to become phosphorylated. Upon doing this, it was found that there were a multitude of residues that were likely to become phosphorylated at the C-terminus end of the protein, which is where I focused further to find conserved residues among different organisms with blood. Using ClustalW2, the SIRT1 protein sequences of various organisms, with and without blood, were aligned and it was found that there were three serine residues (denoted as S) that were conserved among organisms with blood, but were not found in organisms without (figure 4). Furthermore, when looking at the results from NetPhos, it was found that these three residues had a high likelihood of becoming phosphorylated.

Figure 4: The ClustalW2 alignment of the SIRT1 protein from other species. The Serine residues outlined (the S's) are the conserved sites found in those organisms with blood (Humans-Zebrafish) but were not conserved in those without blood (Drosophila-Arabidopsis). The SIRT1 protein in these organisms was also subjected to NetPhos and it was found that these three residues have a high likelihood of becoming phosphorylated. This indicates that these sites might have a role in the activation of SIRT1 in the blood.
Seeing as these residues seem to be specific to organisms
with blood, I would suggest disrupting these sites and observing the resulting effect
on the activity of SIRT1 within the blood. This experiment would consist of
wild type mice (ATM +/+; SIRT1 +/+), leukemic mice without these sites on SIRT1
disrupted (ATM-/-; SIRT1 +/+), and leukemic mice with these specific sites
disrupted, making them, presumably, ATM -/-; SIRT1 -/-. It would be expected
that mice with an ATM-/-; SIRT1 -/- genotype would show a less severe leukemic
phenotype compared to those that are ATM-/-; SIRT +/+. The
reason for this less aggressive leukemic phenotype in ATM -/- and SIRT1 -/-
mice is because SIRT1 is not present to down-regulate p53 and up-regulate
autophagy, which means the leukemic cells are not able to evade apoptosis as
easily, which, in turn, could make them more sensitive to chemotherapeutics. If
these phosphorylation sites were found to be responsible for SIRT1 activation
in the blood, research could then be done to develop small molecules that
inhibited these sites on SIRT1. These molecules could be implemented with
chemotherapy used for CLL patients with an 11q-. Hopefully these
patients would have a more positive response to the treatment, which would allow for better and more efficient care for them.
Below is a link to download the talk that was given presenting the research that was done for this project. If you have any questions, comments or concerns feel free to contact me!
| final_talk_cll_and_atm.pdf |
References:
1. Austen, B., Skowronska, A., Baker, C., Powell, J. E., Gardiner, A., Oscier, D., . . . Stankovic, T. (2007). Mutation Status of the Residual ATM Allele Is an Important Determinant of the Cellular Response to Chemotherapy and Survival in Patients With Chronic Lymphocytic Leukemia Containing an 11q Deletion. Journal of Clincial Oncology, 25(34), 5448-5457. doi:10.1200/JCO.2007.11.2649
2. Skowronska, A., Austen, B., Powell, J. E., Weston, V., Oscier, D. G., Dyer, M. J., . . . Stankovic, T. (2012). ATM germline heterozygosity does not play a role in chronic lymphocytic. Haematologica, 97(1), 142-146. doi:10.3324/haematol.2011.048827
3. Ouillette, P., Li, J., Shaknovich, R., Li, Y., Melnick, A., Shedden, K., & Malek, S. N. (2012). Incidence and Clinical Implications of ATM aberrations in Chronic Lymphocytic Leukemia. Genes,Chromosomes & Cancer, 51(12), 1125-1132. doi:10.1002/gcc.21997
4. Leeuwen, I., & Lain, S. (2009). Sirtuins and p53. Advances in Cancer Research, 102, 171-195. doi:10.1016/S0065-230X(09)02005-3
5. He, L.-q., Lu, J.-h., & Yue, Z.-y. (2013). Autophagy in ageing and ageing-associated diseases. Acta Pharmacol Sin, 34(5), 605-611. doi:10.1038/aps.2012.188
6. Yoon, J.-H., Ahn, S.-G., Lee, B.-H., Jung, S.-H., & Oh, S.-H. (2012). Role of autophagy in chemoresistance: Regulation of the ATM-mediated DNA-damage signaling pathway through activation of DNA–PKcs and PARP-1. Biochemical Pharmacology, 83(6), 747-757. doi:10.1016/j.bcp.2011.12.029
7. Yuan, J., Luo, K., Liu, T., & Lou, Z. (2012). Regulation of SIRT1 activity by genotoxic stress. Genes and development, 26, 791-796. doi:10.1101/gad.188482.112
8. Roth, M., & Chen, W. (2013). Sorting out functions of sirtuins in cancer. Oncogene, 1-12. doi:10.1038/onc.2013.120
9. Firestien, R., Blander, G., Michan, S., Oberdoerffer, P., Ogino, S., Campbell, J., . . . Sinclair, D. A. (2008). The SIRT1 Deacetylase Suppresses Intestinal Tumorgenesis and Colon Cancer Growth. PLOS One, 3(4), e200. doi:10.1371/journal.pone.0002020
10. Hongfeng, Y., Wang, Z., Ling, L., Zhang, H., Modi, H., Horne, D., . . . Chen, W. (2012). Activation of stress response gene SIRT1 by BCR-ABL promotes leukemogensis. Blood, 119(8), 1904-1914. doi:10.1182/blood-2011-06-361691
2. Skowronska, A., Austen, B., Powell, J. E., Weston, V., Oscier, D. G., Dyer, M. J., . . . Stankovic, T. (2012). ATM germline heterozygosity does not play a role in chronic lymphocytic. Haematologica, 97(1), 142-146. doi:10.3324/haematol.2011.048827
3. Ouillette, P., Li, J., Shaknovich, R., Li, Y., Melnick, A., Shedden, K., & Malek, S. N. (2012). Incidence and Clinical Implications of ATM aberrations in Chronic Lymphocytic Leukemia. Genes,Chromosomes & Cancer, 51(12), 1125-1132. doi:10.1002/gcc.21997
4. Leeuwen, I., & Lain, S. (2009). Sirtuins and p53. Advances in Cancer Research, 102, 171-195. doi:10.1016/S0065-230X(09)02005-3
5. He, L.-q., Lu, J.-h., & Yue, Z.-y. (2013). Autophagy in ageing and ageing-associated diseases. Acta Pharmacol Sin, 34(5), 605-611. doi:10.1038/aps.2012.188
6. Yoon, J.-H., Ahn, S.-G., Lee, B.-H., Jung, S.-H., & Oh, S.-H. (2012). Role of autophagy in chemoresistance: Regulation of the ATM-mediated DNA-damage signaling pathway through activation of DNA–PKcs and PARP-1. Biochemical Pharmacology, 83(6), 747-757. doi:10.1016/j.bcp.2011.12.029
7. Yuan, J., Luo, K., Liu, T., & Lou, Z. (2012). Regulation of SIRT1 activity by genotoxic stress. Genes and development, 26, 791-796. doi:10.1101/gad.188482.112
8. Roth, M., & Chen, W. (2013). Sorting out functions of sirtuins in cancer. Oncogene, 1-12. doi:10.1038/onc.2013.120
9. Firestien, R., Blander, G., Michan, S., Oberdoerffer, P., Ogino, S., Campbell, J., . . . Sinclair, D. A. (2008). The SIRT1 Deacetylase Suppresses Intestinal Tumorgenesis and Colon Cancer Growth. PLOS One, 3(4), e200. doi:10.1371/journal.pone.0002020
10. Hongfeng, Y., Wang, Z., Ling, L., Zhang, H., Modi, H., Horne, D., . . . Chen, W. (2012). Activation of stress response gene SIRT1 by BCR-ABL promotes leukemogensis. Blood, 119(8), 1904-1914. doi:10.1182/blood-2011-06-361691
This web page was produced as an assignment for Genetics 677, an undergraduate course at UW-Madison
